Le prion : un agent pathogène peu connu
Il y a quelques décennies, alors que l’agent étiologique était encore inconnu, des maladies neurodégénératives létales et apparemment transmissibles, touchant les humains et les animaux, furent regroupées sous le vocable des encéphalopathies spongiformes transmissibles, ou EST. À défaut de connaître la véritable identité de l’agent responsable, celui-ci fut nommé «agent transmissible non conforme» (ATNC). Ce n’est que dans les années 1980, sous les travaux de l’équipe du professeur Stanley Prusiner, que le caractère très particulier de l’agent étiologique présumé responsable des EST fut confirmé. Les études de ce professeur démontrèrent que des extraits purifiés de cerveaux de hamster, qui présentaient des anomalies à la suite d’une inoculation de la «tremblante», contenaient une glycoprotéine se révélant étroitement liée au pouvoir infectieux. Cette glycoprotéine fut nommée prion (de «proteinaceous infectious particle») et notée «PrP», pour «protease resistant protein». Cette découverte valut au Docteur Prusiner le Prix Nobel de médecine en 1997.
Que sont les prions ?
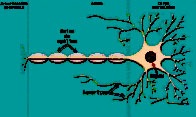
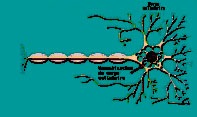
Les prions sont des protéines constituantes naturelles des cellules animales. Pour des raisons encore inconnues, il existe des prions « pathogènes » qui ne sont en fait que des formes altérées des prions normaux. Lorsque ces formes anormales atteignent le cerveau, elles s’accumulent et créent des «plaques amyloïdes» correspondant à des dépôts protéiques dans les espaces intercellulaires. Une lyse des neurones peut ensuite en résulter, créant une vacuolisation dans le cerveau (figure 1).
Figure 1. Encéphalopathie spongiforme bovine
|
Cellule nerveuse saine |
Cellule nerveuse endommagée |
 |
 |
Source: Pillon & Daniel Lepoittevin,L'Encéphalopathie Spongiforme Bovine,
science-citoyen.ustrasbg.fr/dossiers/prion/prion/sympo/esbsym.htlm
Ces mécanismes entraînent une dégénérescence encéphalique donnant lieu à une encéphalopathie dite spongiforme. Cette pathologie se manifeste par une gamme de symptômes neurologiques, locomoteurs, comportementaux et cognitifs qui diffèrent selon l’espèce animale. La maladie à prions la plus connue chez l’humain est la maladie de Creutzfeldt-Jakob (MCJ).
Les protéines prions anormales ont été décrites la première fois dans des cas de tremblante du mouton ou «scrapie» : elles portent le vocable de PrPsc, le sc rappelant son origine dans la maladie «scrapie». D’autre part, la forme normale est notée PrPc, le c rappelant sa provenance cellulaire.
Il est très difficile de dénaturer les PrPsc; ils sont même réfractaires aux méthodes habituelles de désinfection, car ils résistent fort bien à la majorité des méthodes courantes d’inactivation chimique et physique (désinfectants et stérilisation) tout en demeurant stables à une gamme étendue de pH et de températures. Il est donc très difficile de décontaminer des instruments chirurgicaux ayant été utilisés sur des patients atteints de MCJ, ce qui peut poser un sérieux problème de transmission iatrogénique.
La résistance environnementale du PrPsc fait en sorte que la protéine n’est pas inactivée lors de la cuisson ou de la digestion des aliments, permettant, du moins en théorie, la transmission de l’infection par voie alimentaire. Ceci implique que la transmission peut se faire chez certains animaux par le biais des farines carnées ou chez l’homme lors de la consommation de produits alimentaires contaminés par les PrPsc. De plus, ces derniers peuvent demeurer longtemps actifs dans l’environnement, ce qui peut en faciliter la transmission d’un animal à l'autre et augmenter l’importance du réservoir des maladies à prions.
Quelles sont les maladies à prions ?
Les prions sont responsables des EST (encéphalopathies spongiformes transmissibles), maladies dégénératives du système nerveux central, humaines et animales, dont l’évolution est toujours fatale. Ces encéphalopathies ont notamment comme caractéristique d’être transmissibles, du moins en laboratoire, entre un certain nombre d’espèces animales. La description clinique complète de chaque entité pathologique dépasse le propos de ce document et se retrouve dans de nombreuses revues de la littérature traitant du sujet1-8.
Toutes les EST ont des points en commun :
- ce sont des maladies dégénératives du cerveau évoluant généralement sur plusieurs années, quelquefois sur plusieurs mois;
- elles se manifestent par des symptômes locomoteurs de claudication et de tremblement, des problèmes cognitifs, une évolution vers la démence, ou une combinaison de ceux-ci;
- leur diagnostic ne peut être confirmé que post mortem, par histopathologie;
- elles sont probablement associées à des facteurs de susceptibilité génétique;
- elles ont toujours une issue fatale avec une histopathologie comprenant des plaques amyloïdes et l’apparence spongiforme due aux lyses cellulaires.
Les principales maladies à prions reconnues à ce jour chez l’animal sont : la tremblante du mouton; l’encéphalite transmissible du vison (ETV), l’encéphalite spongiforme féline (ESF), la maladie du dépérissement des cervidés (MDC) et l’encéphalite spongiforme bovine (ESB).
Plusieurs formes de maladies à prions existent également chez l’homme :
- la maladie de Creutzfeldt-Jakob, une EST observée chez des personnes âgées ou associée à une transmission iatrogène;
- le Kuru, une EST particulière associée à des rites funéraires dans une région de la Papouasie;
- l’insomnie fatale familiale, une forme rare et héréditaire;
- la maladie de Gerstmann-Sträussler-Scheinker, aussi une forme rare et héréditaire;
- la maladie d’Alpers, possiblement associée à un « agent transmissible non-conforme ».
De plus, à la suite de l'épizootie(a) survenue au milieu des années 1980, il a été démontré que l’ESB pouvait être transmise aux humains par voie alimentaire et engendrer une symptomatologie similaire à celle de la MCJ. Ce syndrome a été appelé nvMCJ (nouvelle variante de la maladie de Creutzfeld-Jakob). Les premiers cas de cette nouvelle forme d’EST sont apparus dix ans après le début de l’épizootie et en 2006, 161 cas étaient dénombrés au total au Royaume-Uni et moins de 30 cas dans les autres pays, principalement européens. Un seul cas de nvMCJ a été rapporté au Canada et il semble qu’il soit associé à la consommation de bœuf lors d’un voyage au Royaume-Uni au moment de l’épizootie.
Chez l’homme, les maladies à prions suivent une évolution lente de plusieurs mois à plusieurs années, mais l’issue fatale survient en quelques semaines à quelques mois après l’apparition des symptômes.
Les prions et la santé publique : impacts et mesures préventives
La plus importante des EST demeure l’épizootie d’ESB qui, avec plus de 170 000 têtes de bétail sacrifiées, des centaines de producteurs dédommagés et des pertes de revenus d’exportation énormes, a coûté au Royaume-Uni plus de 6,4 milliards de livres sterling (£). Comparativement à ces conséquences économiques majeures, l’impact en santé publique pourrait paraître négligeable, particulièrement pour les pays autres que le Royaume-Uni. De fait, l’incidence des EST demeure très faible au Canada et il semblerait que l’application de mesures préventives (dictées par le principe de précaution) et les systèmes de surveillance aussi bien en production animale qu’en médecine humaine aient réussi à contenir la transmission des EST. Toutefois, la transmissibilité inter-espèce des maladies à prions et la résistance de l’agent aux mesures normales de stérilisation en font un risque à la santé humaine.
Épizootie / zoonoses
Malgré la très faible incidence d’ESB au Canada, la présence de prions en production animale est devenue une préoccupation de santé publique étant donné la transmission possible de l’ESB à l’homme par ingestion de produits d’origine animale, provenant de ruminants contaminés. Dans ce contexte, les aliments utilisés pour nourrir le bétail peuvent constituer une source potentielle d’introduction de certains agents pathogènes dans la chaîne alimentaire, dont les prions. Cette préoccupation s’explique dans la mesure où, depuis les années 1970, de nombreuses espèces animales d’élevage reçoivent des aliments qui sont en partie préparés avec des farines de viande et d’os (FVO) parfois appelées farines animales ou farines carnées. Ces dernières sont obtenues à partir de carcasses ou de sous-produits animaux non utilisables pour l’alimentation humaine. Ainsi, dans les premiers mois de l’épizootie, au Royaume-Uni, des carcasses contaminées ont pu être introduites dans la fabrication de farines carnées et peut-être même dans la chaîne alimentaire humaine.
Au Canada, en vertu du Règlement sur la santé des animaux, il est interdit depuis août 1997 de nourrir les ruminants (bœufs, moutons, chèvres, cerfs, wapitis et autres espèces de ce groupe) avec la plupart des protéines de mammifères, à l’exception du lait et de ses dérivés, de la gélatine, du gras ou des produits sanguins. Par contre, toutes les protéines provenant des porcs et des chevaux sont acceptées dans l’alimentation des ruminants.
Par ailleurs, les espèces animales, autres que les ruminants, peuvent être, quant à elles, alimentées avec n’importe quelle protéine de mammifère, incluant les protéines issues des ruminants. Cet état de fait a suscité des craintes au sujet des FVO provenant de résidus de bovins, potentiellement contaminés avec le prion de l’ESB, quant à leur capacité à infecter des animaux comme le porc, chez qui leur utilisation est permise. Cette crainte persiste malgré le fait qu’aucune encéphalopathie porcine n’ait été rapportée et que le Canada soit un pays où l’incidence d’ESB est, rappelons-le, très faible; en août 2007, le programme de surveillance de l’ESB ne rapportait que sept cas depuis son instauration en 1992.
Par ailleurs, il faut ajouter que l’importation de farines d’origine animale produites à l’extérieur du Canada est interdite ou sévèrement réglementée. Selon l’analyse de la littérature scientifique et l’opinion de certains spécialistes consultés, il appert que la probabilité que les porcs soient infectés après avoir consommé des FVO est pratiquement nulle et ne constitue pas un problème préoccupant de santé publique. De plus, une modification apportée à la réglementation en vigueur resserre l’obligation de retirer les matières à risque spécifiées, soit les mêmes matières que celles retirées du bétail abattu pour la consommation humaine, avant transformation. Dorénavant, l’usage de ces matières est interdit dans la composition des aliments pour animaux, y compris ceux destinés aux animaux domestiques, ainsi que dans les engrais.
Maladies iatrogènes
La MCJ ne peut se transmettre de personne à personne, par simples contacts, mais elle le peut par la transplantation de tissus infectés, par la prise d’hormones pituitaires contaminées ou par manipulations effractives avec des instruments chirurgicaux ou sondes contaminées. Ces dernières possibilités imposent des mesures très strictes de contrôle des infections lorsque des manipulations à risque doivent être effectuées sur des sujets soupçonnés être atteints d'une maladie à prions. Ces mesures de précautions exceptionnelles s’appliquent à la salle d’opération, à la salle d’autopsie, au laboratoire de pathologie et à la salle de thanatologie. Les précautions extrêmement exigeantes sont dictées par la résistance environnementale des prions.
La possibilité de confirmer de manière rapide et fiable l’infection d’une personne en début de maladie constitue un élément clé de la prévention et du contrôle des maladies infectieuses en général. Dans le cas des ESB, les maladies sont précédées d’une longue «période d’incubation», cliniquement silencieuse, pendant laquelle l’individu infecté peut théoriquement transmettre la maladie à d’autres personnes. Face à cette situation, des recherches appliquées s’imposent afin d’améliorer la vitesse et l’exactitude des méthodes de diagnostic pour déceler les agents et les maladies à prions.
Système national de surveillance
Créé à titre de système national de surveillance par Santé Canada en 1998, le SS-MCJ entretient une surveillance active de la maladie de Creutzfeldt-Jakob sous la responsabilité de l’Agence de la santé publique du Canada. Le SS-MCJ applique des critères élaborés par l’Organisation mondiale de la santé lorsqu’il établit un diagnostic de cas possible, probable ou certain de MCJ. Les rapports de cas transmis au système ont été relativement constants. Cela est probablement dû à une sensibilisation soutenue à la maladie, à une déclaration continue par les médecins et à l’accès au dépistage de la protéine 14-3-3 (test efficace du diagnostic de la MCJ) offert par le système de surveillance.
Perspectives
Certains épidémiologistes avaient envisagé des scénarios plutôt pessimistes quant à l’incidence probable de la nvMCJ dans les prochaines décennies (plusieurs milliers de victimes au Royaume-Uni) étant donné la prévalence de l’exposition et l’apparente absence de facteurs prédisposant. Avec le recul, ces prédictions ont été contredites par une incidence observée de moins de 170 cas sur 10 ans et par l’absence apparente de nouveaux cas depuis 2006.
Enfin, bien que les autorités de santé publique soient en quelque sorte rassurées, certains craignent encore que l’incubation de la nvMCJ, puisse dépasser une période de 50 ans, tel qu’observé pour certains cas sporadiques de MCJ chez les personnes âgées ou pour les 11 cas récents de Kuru identifiés par l’équipe de Collinge, qui seraient devenus symptomatiques entre 1996 et 2004, soit 40 ans après la cessation des rites funéraires à risque.
Conclusion
Il n’existe actuellement aucun traitement efficace contre les EST humaine ou animale, bien que plusieurs agents chimiques et immunologiques aient été proposés à la suite d’expériences en laboratoire pour modifier l’évolution et/ou la durée de la maladie. Par ailleurs, il n’existe aucun traitement préventif, telle la vaccination, par exemple. La mise au point d’agents préventifs et thérapeutiques demeure un objectif à long terme. Pour protéger la santé des animaux et des humains, les seules interventions s’offrant présentement à la santé publique sont l’amélioration de la salubrité des aliments et les mesures spécifiques de traitement de toute matière ou objet potentiellement contaminés. De plus, il est mondialement reconnu qu’il est nécessaire d’instaurer des mécanismes efficaces de surveillance des ESB/EST et de suivi des animaux, et de pouvoir répondre aux nouveaux événements et problèmes, lesquels, comme le démontre l’expérience passée avec les épizooties, pourraient se reproduire.
Comme le Canada a maintenant connu des occurrences de toutes les principales formes des EST chez le bétail domestique, la faune et les humains et que l’on sait que les maladies peuvent se transmettre entre les espèces, il est essentiel de les étudier de manière intégrée, soit sous l’angle de la prévention et du contrôle des maladies, soit sous celui des impacts économiques et sociaux9.
Références
- Aguzzi, A. and Polymenidou, M. (2004). Mammalian prion biology: one century of evolving concepts. Cell, 116 : 313-27.
- Dormont, D. (1994). [Natural history of human transmissible subacute spongiform encephalopathies]. Transfus Clin Biol, 1 : 319-31.
- Lasmezas, C. I., Fournier, J. G., Nouvel, V., Boe, H., Marce, D., Lamoury, F., Kopp, N., Hauw, J. J., Ironside, J., Bruce, M., Dormont, D., and Deslys, J. P. (2001). Adaptation of the bovine spongiform encephalopathy agent to primates and comparison with Creutzfeldt-Jakob disease: implications for human health. Proc Natl Acad Sci U.S.A, 98 :4142-7.
- Dormont, D. (2000). New variant of Creutzfeldt-Jakob disease. Eurosurveillance, 5 : 95-7.
- Donnelly, CA, Gore, S. M., Curnow, R. N., and Wilesmith, J. W. (2006). Surveillance de la tremblante. Contribuez à éradiquer la tremblante au Canada. Site de l’Agence canadienne d’inspection des aliments,
- Marsh, R. F. and Bessen, R. A. (1993). Epidemiologic and experimental studies on transmissible mink encephalopathy. Dev Biol Stand , 80 : 111-8.
- Ricketts, M. N. (2004). Public health and the BSE epidemic. Curr Top Microbiol Immunol., 284 : 99-119.
- Prusiner, S. B. (1998). Prions. Proc Natl Acad Sci U.S.A, 95 : 13363-83.
- Santé Canada et Réseaux de centres d’excellence.(2006). Priorités de recherche sur les encéphalopathies spongiformes transmissibles et les encéphalopathies bovines spongiformes. Annexe A.
(a) Maladie qui frappe simultanément un grand nombre d’animaux de même espèce ou d’espèces différentes.