Mécanismes d’action et de toxicité de l’acétaminophène
Volume 27, Numéro 1
Introduction
L’acétaminophène (paracétamol, APAP) est un médicament faisant partie de la classe des antalgiques antipyrétiques non salicylés. Il possède des propriétés analgésiques, antipyrétiques et même antioxydantes(1-3). Il est indiqué dans le traitement symptomatique de la fièvre et des douleurs d'intensité faible à modérée. Contrairement aux anti-inflammatoires non stéroïdiens (AINS) tels que l’acide acétylsalicylique ou l’ibuprofène, l’APAP ne possède pas de propriétés anti-inflammatoires et n'agit pas sur l'agrégation plaquettaire(2).
S’il est clair que l’APAP agit au niveau du système nerveux central (SNC), son mécanisme d'action complet n’est toutefois pas encore totalement élucidé, et ce, en dépit du fait que cette molécule fut synthétisée pour la première fois il y a plus de 120 ans(4).
Les cyclooxygénases (COX) font partie d'un complexe enzymatique convertissant l'acide arachidonique en prostaglandine H2 (PGH2)(5). Les AINS comme l’ibuprofène et l’acide acétylsalicylique, ayant des propriétés anti-inflammatoires et antiplaquettaires, sont également des inhibiteurs non spécifiques des COX-2(6,7). Par contre, l’APAP est considéré par certains comme un faible inhibiteur de la synthèse des prostaglandines (PG) par l’entremise des COX-2. En effet, des études ont démontré que l’APAP serait plus sélectif aux COX-2 qu’aux COX-1(8,9). L’hypothèse proposée serait que l’APAP n’inhiberait pas les enzymes COX dans un milieu riche en radicaux peroxydes, ce qui est le cas des zones d’inflammations(10). Cependant, cette hypothèse n’explique pas pourquoi l’APAP ne possède pas d’effets antiplaquettaires.
Une théorie, qui n’a pas encore été confirmée chez l’humain, stipulant que l’APAP lierait possiblement un autre type de COX (COX-3), pourrait expliquer pourquoi l’APAP réduit la fièvre et la douleur tout en n’ayant aucune activité anti-inflammatoire et antiplaquettaire(11,12). Cependant, comme plusieurs études ont tenté, en vain, de prouver cette théorie chez l’homme, cette dernière semble de moins en moins probable(8).
Une étude, publiée en 2005, a tenté d'expliquer le mécanisme d'action d'une toute autre façon. Selon cette étude, l'APAP serait métabolisé en p-aminophénol au niveau du cerveau. Le p-aminophénol serait par la suite conjugué à l'acide arachidonique pour former de la N-arachidonyl-phénolamine (AM404). C’est cette molécule qui serait pharmacologiquement active au cerveau en inhibant la synthèse de prostaglandines(13,14).
Une autre hypothèse explique que l’APAP exercerait son effet analgésique au niveau du SNC par une potentialisation des neurones sérotoninergiques descendants de la moelle épinière, ceci ayant pour effet d’exercer un contrôle inhibiteur sur les voies nociceptives(15).
Pharmacocinétique(2,16)
Entre 60 et 98 % d’une dose d’APAP est rapidement absorbée, au niveau de l'intestin grêle, après ingestion orale. La concentration plasmatique maximale est atteinte en 15 minutes pour les comprimés à libération immédiate et entre 30 et 60 minutes pour les comprimés à libération prolongée. La prise simultanée de comprimés d’APAP et de nourriture diminue significativement (près de 50 %) la concentration maximale atteinte au niveau sanguin.
L’APAP se distribue rapidement dans la plupart les tissus. Entre 10 et 30 % d’une dose normale se liera aux protéines plasmatiques telles que l’albumine, contrairement à 20 à 50 % lors d’un surdosage. Les concentrations d’APAP mesurées dans le sang, la salive et le plasma sont relativement comparables. Le volume de distribution de ce médicament se situe entre 0,7 et 1 L/kg chez l’enfant et entre 1 et 2 L/kg chez l’adulte. L’APAP est reconnu pour traverser les barrières placentaire et hématoencéphalique.
Le métabolisme de l’APAP se produit essentiellement au niveau du foie. Près de 25 % d’une dose normale ingérée per os est métabolisée lors du premier passage hépatique. Les deux voies métaboliques principales étant impliquées dans le métabolisme de l’APAP sont la glucuroconjugaison (~60 %) et la sulfoconjugaison (~25 %). Cependant, une troisième voie métabolique est également sollicitée (~12 à 15 %). Elle est catalysée par les enzymes CYP2E1, CYP1A2 et aussi CYP3A4 du cytochrome p450. Grâce à cette voie métabolique, l’APAP est métabolisé par désacétylation et par N-hydroxylation. Le dérivé N-hydroxylé (4 %) est un intermédiaire toxique que l’on nomme N-acétyl p-benzoquinone imine ou NAPQI. À dose thérapeutique d’APAP, le NAPQI est rapidement neutralisé par une réaction avec le glutathion, puis éliminé dans l’urine à la suite d’une réaction de conjugaison avec la cystéine et l'acide mercapturique. Lors d’intoxication à l’APAP, cette voie métabolique gagne en importance et dans les cas où les concentrations en glutathion deviennent insuffisantes pour neutraliser le NAPQI, celui-ci peut générer sa toxicité. Les patients souffrant d’alcoolisme chronique, malnutrition, anorexie, boulimie, VIH, fibrose kystique ou jeûne prolongé ont généralement des concentrations de glutathion endogène plus faibles que la normale et sont plus susceptibles de réagir négativement à de fortes doses d’APAP. Finalement, une quatrième voie mineure aboutissant en 3-hydroxyparacétamol doit également être considérée.
De la naissance à 2 ans, la métabolisation de l’APAP s’effectue également par les enzymes CYP2E1. Cependant, un pourcentage plus élevé de la dose administrée est excrété sous forme de conjugué sulfonide, comparé à chez l’adulte. Ainsi, l’utilisation plus importante de cette voie compenserait la déficience de la voie de métabolisation par glucuroconjugaison, chez cette jeune population(17).
Figure 1 - Résumé des voies métaboliques de l’acétaminophène
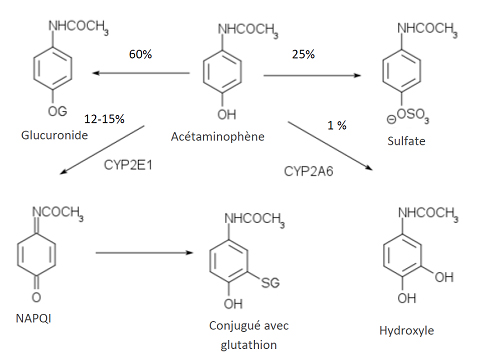
L'élimination de l’APAP et de ses métabolites est essentiellement urinaire : 90 % de la dose ingérée est éliminée par le rein en 24 heures, dont moins de 5 % est éliminée sous forme inchangée. La demi-vie d'élimination varie généralement entre 1 à 3 heures, mais celle-ci peut être augmentée de 2 à 4 heures chez les personnes souffrant d’insuffisance hépatique sévère, et de 4 à 6 heures chez les personnes en surdosage.
Toxicité de l’acétaminophène
L’intervalle thérapeutique de l’APAP se situe entre 65 et 130 µmol/L(18,19).
Trois mécanismes d’action reliés à la toxicité de l’APAP sont proposés :
- Intoxication aigüe qui surcharge le métabolisme hépatique;
- Activation excessive des cytochromes pouvant mener, entre autres, à des interactions médicamenteuses;
- Faible présence, avant l’exposition, de quantités endogènes de glutathion soit à cause d’un facteur génétique, d’une intoxication, d’une malnutrition ou de l’alcoolisme.
Comme mentionné ci-dessus, l’APAP n’est pas considéré comme toxique, en revanche, un de ses métabolites, le N-acétyl p-benzoquinone imine (NAPQI), l’est. À dose thérapeutique, le NAPQI est rapidement conjugué avec le glutathion afin d’être détoxiqué et par la suite, éliminé dans l’urine. Cependant, à fortes doses, les réserves en glutathion baissent significativement, tout comme l’effet protecteur de ce dernier sur le NAPQI. La toxicité étant fonction de la dose, l’ingestion de fortes doses d’APAP est donc susceptible d’induire une insuffisance hépatique aigüe.
L’adduction du NAPQI aux protéines cellulaires a pour conséquence de modifier les structures et les fonctions de ces dernières. Ainsi, le NAPQI perturbe l'homéostasie calcique, génère une perte d’ATP et de l’œdème cellulaire, produit des radicaux réactifs de l’oxygène et de nitrotyrosine avec peroxydation lipidique. On assiste ensuite à une augmentation de la perméabilité des mitochondries, qui entraînera rapidement la mort cellulaire. Ceci génère ainsi une nécrose centrolobulaire et finalement une hépatite cytolytique. Une atteinte rénale découlant du même mécanisme d’action est également possible. Dans un foie sain, l'antidote N-acétylcystéine permet de restaurer les quantités de glutathion utilisées.
Présentation clinique
Les nombreux effets indésirables reliés à une exposition aigüe à l’APAP sont séparés en 4 stades différents étalés, en moyenne, sur une période de 5 jours(20).
| Stades | Temps postingestion (h) | Effets observés |
|---|---|---|
| I |
< 24 |
Asymptomatique, salivation, nausées, vomissements, anorexie (enfants surtout), pâleur, diaphorèse, faiblesse, somnolence. Début de l’augmentation des taux de AST et ALT (> 12 heures postingestion) |
| II |
24 à 72 |
Résolution des symptômes initiaux. Douleurs à l’hypocondre droit, tachycardie, hypotension, nausées, vomissements, anorexie. Poursuite de l’augmentation des taux d’AST et d’ALT et, dans des intoxications sévères, des taux de bilirubine et de prothrombine. |
| III |
72 à 96 |
Douleur à l’hypocondre droit, vomissements, insuffisance hépatique et/ou rénale, pancréatite, jaunisse, coagulopathie, hypoglycémie, encéphalopathie hépatique, acidose métabolique. Atteinte des taux maximaux en AST, ALT, bilirubine et en prothrombine. |
| IV |
> 120 |
Résolution complète des symptômes et des insuffisances |
Conclusion
Isolé pour la première fois il y a plus de 120 ans, l’APAP soulève encore plusieurs interrogations concernant l’ensemble de sa pharmacodynamie, qui n’est pas encore totalement élucidée. Cependant, la pharmacocinétique de cette molécule ainsi que la toxicité générée par l’un de ses métabolites, le NAPQI, sont en revanche très bien documentées.
Pour toute correspondance
Pierre-Yves TremblayInstitut national de santé publique du Québec
945, avenue Wolfe, 4e étage, Québec (Québec) G1V 5B3
Téléphone : 418 650-5115, poste 4653
Télécopieur : 418 654-2148
Courriel : [email protected]
Références
1Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis 2007 Aug;11(3):525-48, vi.
2Acetaminophen in DRUGDEX Evaluations. Micromedex Healthcare Series Online 2010-11-18; [En ligne] http://www.thomsonhc.com (consulté le 2010-12-08).
3Anderson BJ. Paracetamol (Acetaminophen): mechanisms of action. Paediatr Anaesth 2008 Oct;18(10):915-21.
4Anon. Pain relief: From coal tar to paracetamol. Education in Chemistry 2005 Jul;42(4).
5Foegh M, Ramwell P. The Eicosanoids: Prostagladins, Thromboxanes, Leukotrienes & Related Compounds. In: Mcgraw Hill, editor. Basic & Clinical Pharmacology. 2004. p. 298-312.
6Rainsford KD. Ibuprofen: pharmacology, efficacy and safety. Inflammopharmacology 2009 Dec;17(6):275-342.
7Autacoids: Drug Therapy of Inflammation, Chapter 26: Analgesic-Antipyretic and Antiinflammatory Agents; Pharmacotherapy of Gout. In: Mcgraw Hill, editor. The Pharmacological Basis of Therapeutic. 11 ed. 2006. p. 671-715.
8Hinz B, Cheremina O, Brune K. Acetaminophen (paracetamol) is a selective cyclooxygenase-2 inhibitor in man. FASEB J 2008 Feb;22(2):383-90.
9Graham GG, Scott KF. Mechanism of action of paracetamol. Am J Ther 2005 Jan;12(1):46-55.
10Casimir N, Antignac M, Farihotti R. Antalgiques Non-Opiacés. In: Le moniteur, editor. Médicaments 3e Édition. 2007. p. 395-412.
11Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS, Simmons DL. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A 2002 Oct 15;99(21):13926-31.
12Rezende RM, Franca DS, Menezes GB, dos Reis WG, Bakhle YS, Francischi JN. Different mechanisms underlie the analgesic actions of paracetamol and dipyrone in a rat model of inflammatory pain. Br J Pharmacol 2008 Feb;153(4):760-8.
13Hogestatt ED, Jonsson BA, Ermund A, Andersson DA, Bjork H, Alexander JP, Cravatt BF, Basbaum AI, Zygmunt PM. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem 2005 Sep 9;280(36):31405-12.
14Bertolini A, Ferrari A, Ottani A, Guerzoni S, Tacchi R, Leone S. Paracetamol: new vistas of an old drug. CNS Drug Rev 2006;12(3-4):250-75.
15Tjolsen A, Lund A, Hole K. Antinociceptive effect of paracetamol in rats is partly dependent on spinal serotonergic systems. Eur J Pharmacol 1991 Feb 7;193(2):193-201.
16Baselt R. Acetaminophen. Disposition of toxic drugs and chemicals in man. 6 ed. Foster city: Biomedial Publication; 2002. p. 5-9.
17Tanaka E. In vivo age-related changes in hepatic drug-oxidizing capacity in humans 1. J Clin Pharm Ther 1998 Aug;23(4):247-55.
18Hammett-Stabler CA, Dasgupta A, American Association for Clinical Chemistry. Therapeutic drug monitoring data a concise guide. 3rd ed. Washington, DC: AACC Press; 2007.
19TIAFT 2009. Reference blood level list of therapeutic and toxic substances. 9-24-2004. Ref Type: Internet Communication
20Porter R, Kaplan J. Acetaminophen poisoning. Merck Manuals Online Medical Library 2009-04; [En ligne] http://www.merckmanuals.com/professional/sec21/ch226/ch226c.html
Tremblay PY. Mécanismes d'action et de toxicité de l'acétaminophène. Bulletin d'information toxicologique 2011;27(1). [En ligne] https://www.inspq.qc.ca/toxicologie-clinique/mecanismes-d-action-et-de-…
Bulletin d'information toxicologique, Volume 27, Numéro 1, janvier 2011
La reproduction du contenu du Bulletin d’information toxicologique est autorisée à condition d'en mentionner la source. Toute utilisation à des fins commerciales ou publicitaires est cependant strictement interdite. Les articles publiés dans ce bulletin n'engagent que la responsabilité de leurs auteurs et non celle de l'INSPQ ou du CAPQ.
ISSN : 1927-0801